The Journal of Clinical and Preventive Cardiology has moved to a new website. You are currently visiting the old
website of the journal. To access the latest content, please visit www.jcpconline.org.
Cardiovascular Disease in Patients with Type 2 Diabetes Mellitus
Volume 2, Oct 2013
G. N. Viswanathan, A. G. Zaman, Newcastle upon Tyne, UK
J Clin Prev Cardiol. 2013;2(4):185-9
The prevalence of patients with diabetes mellitus is rising at an exponential rate globally with type 2 diabetes mellitus (T2DM) is the most common form. The presence of coronary artery disease (CAD) in patients with T2DM results in significant mortality and morbidity. The relative risk of cardiovascular events in T2DM remains unchanged despite good control of diabetes mellitus and other cardiovascular risk factors. Recurrent cardiovascular thrombotic events are more common in patients with CAD and T2DM despite current “gold standard” secondary prevention therapy (1). Blood thrombogenicity (ability of blood to clot or “vulnerable blood”) remains higher in T2DM and is associated with poorer outcomes. Better understanding and identification of novel therapeutic targets to reduce thrombogenicity may improve clinical outcomes in this high-risk population.
Epidemiology of Diabetes Mellitus
It is estimated that there will be 4 million people in the UK with diabetes by 2025 and the majority will have T2DM. The impact of diabetes mellitus and its comorbidities on healthcare resources is profound and currently 10% of total healthcare expenditure in UK is spent on care of patients with diabetes (www.diabetes.org.uk, accessed 3/2/2012). The challenges in managing individual patients with diabetes are sizeable and the impact on healthcare resources is likely to exceed current predictions.
Cardiovascular Mortality and Morbidity in T2DM
Diabetes accounts for 6% of total global mortality, Newcastle upon Tyne, UKhalf of the diabetes-associated deaths attributed to cardiovascular disease (CVD) (http://www.idf.org/home/index, accessed 14/02/2011). Diabetes mellitus reduces life expectancy by a decade and it is generally accepted that 80% of patients with T2DM will develop and possibly die of macrovascular disease, predominantly due to cardiac causes. (http://diabetes.niddk.nih.gov/dm/pubs/statistics, accessed 14/2/2012). Haffner and coworkers have shown that diabetic patients without a prior history of myocardial infarction (MI) carried the same risk of death as nondiabetic patients with a previous MI. This has led to diabetes being called a CVD equivalent. However, in the context of aggressive modern secondary prevention strategies, some authors have questioned this assumption (2).
In the presence of diabetes, the incidence of MI is doubled and survival rates are lower. Although cardiovascular mortality has declined over the last decade both in patients with and without diabetes, the relative risk between patients with and without diabetes remains unchanged, with the former suffering a 2.4-fold increase in cardiovascular mortality despite optimal evidence-based primary and secondary prevention strategies (3).
Individuals with diabetes are twice as likely to be admitted to hospital. In a UK hospital admissions study, at any given time, nearly 10% of all in-patients had diabetes and experienced prolonged stays in hospital, resulting in about 80,000 additional bed days per year in the UK. In addition, diabetes accounts for 2.4 times higher medical expenditure compared to a similar population in the absence of diabetes (4). Nevertheless, it is recognized that the most effective way of reducing the “spiraling trend” of medical expenditure in patients with T2DM is to optimize strategies of prevention of cardiovascular events (5).
Platelet dysfunction is present in patients with T2DM. The platelets respond to sub-threshold stimuli and are constantly in activation even in the absence of a major plaque event and have been labeled as “angry platelets” (10). Platelet leucocyte interaction is responsible for propagation and stabilization of thrombus. T2DM is associated with higher platelet leucocyte linkage mediated by P-selectin and cytokines and release of procoagulant, oxidative and mitogenic stimulants, thereby playing a significant role in stabilization of arterial thrombus and capillary microembolism and arterial thrombosis (Table 1).

Coagulation factor abnormalities are common in T2DM. In addition to conventional screening tests such as shorter prothrombin and partial thromboplastin times (PT and APTT), T2DM is associated with elevated tissue factor, reduced tissue factor pathway inhibitor, higher prothrombin activating factor and higher thrombin–antithrombin complex. Increased levels of factor VIII, plasma fibrinogen and Von Willebrand factor and factor XI found in T2DM also promote thrombus formation. In T2DM, reduced fibrinolysis potentiates thrombogenicity and also promotes atherogenesis by extended fibrin deposition. PAI-1, the most potent inhibitor of the fibrinolytic system, is consistently elevated in the vessel walls of patients with T2DM (11) .
Inflammation is associated with arterial, venous and microvascular thrombosis and it has been widely accepted that inflammation up-regulates procoagulants and downregulates anticoagulants and fibrinolysis. Tumor necrosis factor-alpha (TNFα) levels are higher in T2DM and it has been linked to insulin resistance, thrombotic events and microvascular complications seen in T2DM. Suppression of TNFα either by biological antagonists or weight reduction and exercise have been shown to improve glycemic control in T2DM (11). It is plausible but remains to be proven that similar benefits can be achieved in alleviating the prothrombotic state in T2DM by pharmacological inhibition of cytokines.
Coronary Artery Disease in T2DM
Atherothrombotic disease is responsible for significant mortality and morbidity with widespread changes in coronary arteries, despite good control of diabetes mellitus and other cardiovascular risk factors. The pathophysiological mechanisms contributing to CAD in T2DM are multifactorial, complex and interlinked. Contrary to earlier belief that CAD in diabetes is secondary to hyperglycemia, multiple recent epidemiological studies and clinical followup studies strongly suggest that factors other than hyperglycemia play a significant role in the development of CAD in T2DM (6).
Every stage in the process of coronary atherosclerosis namely initiation, progression and plaque rupture is widespread and accelerated in T2DM. Coronary atherosclerotic burden is consistently higher in medium sized arteries in T2DM, along with microvascular disease. Besides, atherosclerotic changes are more dynamic from initiation to progression in T2DM, resulting in early plaque disruption and thus heralding more frequent occlusive atherothrombotic events. In addition, mild to moderate sized atherosclerotic plaques known as “non-flow limiting coronary atherosclerotic lesions” occur more frequently in T2DM and are associated with the majority of the plaque rupture events and thrombosis resulting in non-ST elevation acute coronary syndrome (NSTE-ACS) in T2DM (7).
Excess atheroma burden in patients with T2DM is attributed to loss of the balance in the endothelial damage repair cycle. In the presence of frequently associated cardiovascular risk factors in T2DM such as hypertension, dyslipidemia and obesity, damage to endothelium is more pronounced and repair mechanisms are rapidly exhausted, thereby resulting in early and widespread atheromatous disease (8). Atherosclerotic plaques in T2DM are more prone to rupture (more in number, thinner fibrous caps, increase in inflammatory cells) and have been labeled as “vulnerable plaque.” Other factors can also increase plaque vulnerability such as increased shear stress of flowing blood in patients with T2DM and higher levels of inflammatory mediators such as cytokines and mononuclear phagocytes resulting in weakening of the protective fibrous cap (9).
Blood Thrombogenicity in T2DM
Thrombus formation is a protective mechanism by which hemostasis is maintained in a closed, high-pressure circulatory system after vascular damage. A balance exists between pathways that initiate thrombus formation and its dissolution. A highly specialized and efficient hemostatic system consisting of cellular (platelets) and noncellular (coagulation factors) components is responsible for the three main components of thrombosis as follows:
- Initiation of thrombosis
- Propagation of thrombus
- Autolysis of thrombus
Enhanced thrombogenicity (“vulnerable blood”) seen in T2DM is secondary to changes in all three components by virtue of among others:
- Hyperactive platelets,
- Higher levels of clotting factors and
- Impaired fibrinolysis thereby resulting in T2DM being called as “prothrombotic state.”
Platelet dysfunction is present in patients with T2DM. The platelets respond to sub-threshold stimuli and are constantly in activation even in the absence of a major plaque event and have been labeled as “angry platelets” (10). Platelet leucocyte interaction is responsible for propagation and stabilization of thrombus. T2DM is associated with higher platelet leucocyte linkage mediated by P-selectin and cytokines and release of procoagulant, oxidative and mitogenic stimulants, thereby playing a significant role in stabilization of arterial thrombus and capillary microembolism and arterial thrombosis (Table 1).
Coagulation factor abnormalities are common in T2DM. In addition to conventional screening tests such as shorter prothrombin and partial thromboplastin times (PT and APTT), T2DM is associated with elevated tissue factor, reduced tissue factor pathway inhibitor, higher prothrombin activating factor and higher thrombin–antithrombin complex. Increased levels of factor VIII, plasma fibrinogen and Von Willebrand factor and factor XI found in T2DM also promote thrombus formation. In T2DM, reduced fibrinolysis potentiates thrombogenicity and also promotes atherogenesis by extended fibrin deposition. PAI-1, the most potent inhibitor of the fibrinolytic system, is consistently elevated in the vessel walls of patients with T2DM (11) .
Inflammation is associated with arterial, venous and microvascular thrombosis and it has been widely accepted that inflammation up-regulates procoagulants and downregulates anticoagulants and fibrinolysis. Tumor necrosis factor-alpha (TNFα) levels are higher in T2DM and it has been linked to insulin resistance, thrombotic events and microvascular complications seen in T2DM. Suppression of TNFα either by biological antagonists or weight reduction and exercise have been shown to improve glycemic control in T2DM (11). It is plausible but remains to be proven that similar benefits can be achieved in alleviating the prothrombotic state in T2DM by pharmacological inhibition of cytokines.
Contemporary Studies of Whole Blood Thrombogenicity in Patients with T2DM
Recurrent cardiovascular events are commoner in patients with T2DM and recurrent subclinical thrombotic events may also accelerate atheroma progression. More detailed knowledge about blood thrombogenicity in T2DM can help clinicians optimize antithrombotic therapy in this high-risk group. Better understanding of thrombogenicity in T2DM may help to identify novel therapeutic targets in the diabetic population.
- After NSTE-ACS (Study 1) and
- Stable CAD (Study 2)
The hypothesis for Study 1 (ACS patients) was that patients with T2DM have increased thrombus following NSTE-ACS, despite recommended secondary prevention therapy (www.clinicaltrials.gov, NCT00728286; www.ukcrn.org.uk, 7338).
The hypothesis for Study 2 (stable CAD patients) was that addition of clopidogrel to standard aspirin treatment would be ineffective in reducing thrombus in patients with T2DM and CAD (www.clinicaltrials.gov, NCT00728156; www.ukcrn.org.uk. 5159; Eudract number: 2006-003745-16; MHRA number: 31088/0006/001-0001).
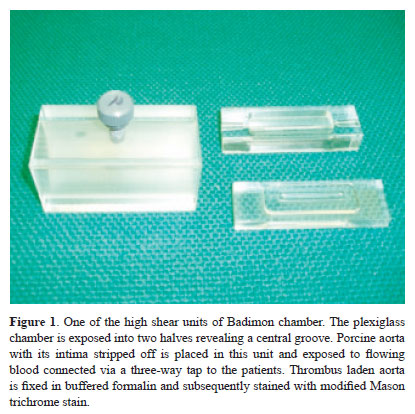
The Badimon chamber (Fig.1), a validated ex vivo arterial injury model was used. This model simulates the in vivo situation of high shear arterial wall damage and quantifies platelet-dependent thrombus, which is the endpoint of all in vitro hemostatic abnormalities (12).
References
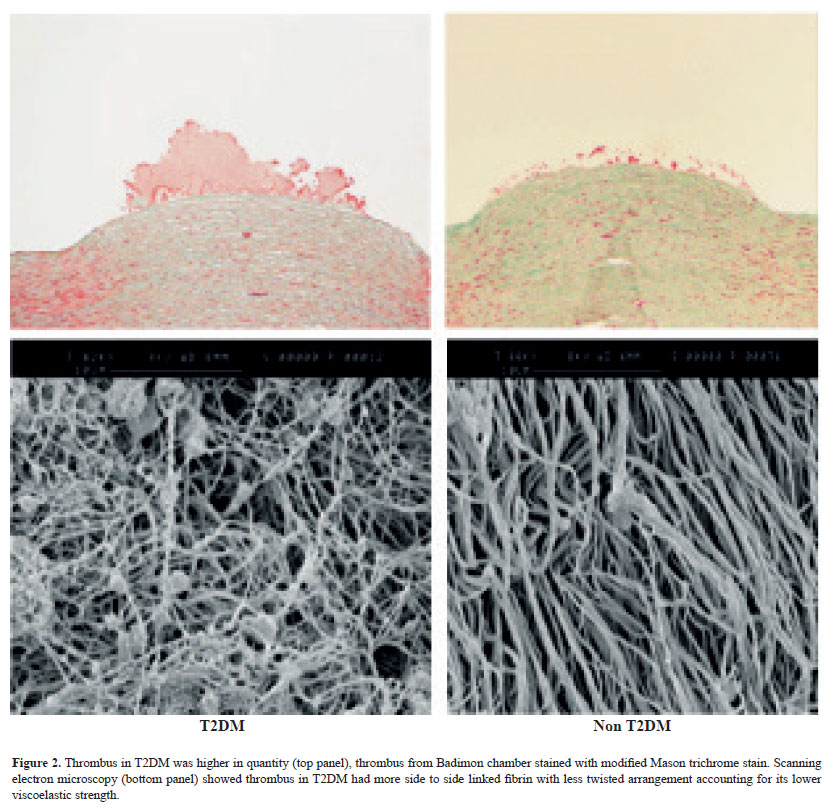
In patients presenting with NSTE-ACS thrombus was significantly increased in patients with T2DM despite evidence-based optimal secondary prevention therapy. Looking at the formed thrombus under electron microscopy a marked difference in clot structure was seen (Fig. 2). Fibrin strands had an irregular pattern with cross-linking in the T2DM population. Using thromboelastography to assess the rate of thrombus lysis, a significantly slower rate of lysis was seen in patients with T2DM. Thrombus kinetics were impaired when studied using thromboelastography with T2DM patients having lower viscoelastic tensile strength [clot index: median (IQR), -0.2(-1.7 to 0.7) vs. 1.0(-0.9 to 3.3), p=0.04] and impaired autolysis [rate of thrombus retraction mm/min: median (IQR), 27.8(11.7–70.7) vs. 78.8(68.5–109.6) p<0.05)]. There was significant but moderate correlation between autolysis and thrombus quantity (rho 0.450, p=0.02) (Unpublished data, presented at Annual Professional Conference, Diabetes UK, 2010). The study revealed higher thrombus burden in T2DM patients [thrombus area, μ2 per mm in T2DM (mean±SD) 20,414±12,344 vs. non-DM 14,933±8,415 p=0.023, 95% CI 811–10674] (Unpublished data, presented in Scientific Sessions, American Heart Association, 2010, Chicago, IL].
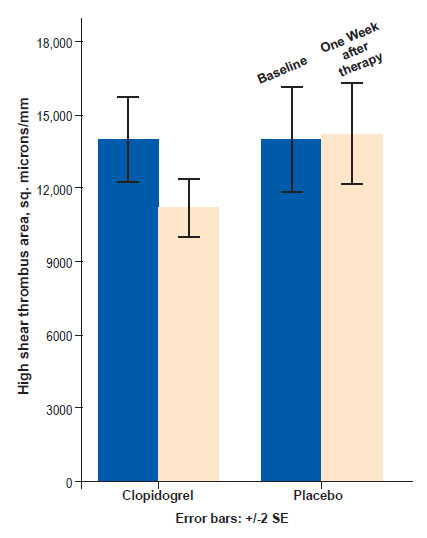
In patients with stable CAD a randomized controlled study of the addition of clopidogrel or placebo to aspirin revealed a significant reduction in thrombus area (Fig. 3). This suggests that the addition (to aspirin) of antiplatelet agents in patients with stable CAD and T2DM is effective at reducing thrombus.
Conclusions
Patients with T2DM and CAD suffer from poorer outcomes despite current aggressive prevention strategies. Blood thrombogenicity (“vulnerable blood”) is higher in these individuals despite optimal antiplatelet therapy. Modification of the vulnerable blood seen in this population by addition of more or different antithrombotic agents or medications to target different aspects of the coagulation pathway may improve cardiovascular mortality. With the global pandemic of T2DM coupled with the failure of large trials of aggressive risk factor control to reduce macrovascular complications in the diabetic population, the need to study mechanisms that target thrombosis and reduce atherothrombotic complications is paramount.
References
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diab Care. 2004;27:1047–53.
- Bulugahapitiya U, Siyambalapitiya S, Sithole J, Idris I. Is diabetes a coronary risk equivalent? Systematic review and meta-analysis. Diab Med. 2009;26:142–8.
- Preis SR, Hwang S-J, Coady S, et al. Trends in all-cause and cardiovascular disease mortality among women and men with and without diabetes mellitus in the Framingham Heart Study, 1950 to 2005. Circulation. 2009;119:1728–35.
- Sampson MJ, Crowle T, Dhatariya K, et al. Trends in bed occupancy for inpatients with diabetes before and after the introduction of a diabetes inpatient specialist nurse service. Diab Med. 2006;23:1008–15.
- Moore P. Type 2 diabetes is a major drain on resources. BMJ. 2000;320:732.
- Meier M, Hummel M. Cardiovascular disease and intensive glucose control in type 2 diabetes mellitus: moving practice toward evidence-based strategies. Vasc Health Risk Manag. 2009;5:859–71.
- Virmani R, Burke AP, Kolodgie F. Morphological characteristics of coronary atherosclerosis in diabetes mellitus. Can J Cardiol. 2006;22:81B–84B.
- Moreno PR, Fuster V. New aspects in the pathogenesis of diabetic atherothrombosis. J Am Coll Cardiol. 2004;44:2293–300.
- Hess K, Grant PJ. Inflammation and thrombosis in diabetes. Thromb Haemost. 2011;105(Suppl 1):S43–54.
- Bhatt DL. What makes platelets angry: diabetes, fibrinogen, obesity, and impaired response to antiplatelet therapy? J Am Coll Cardiol. 2008;52:1060–1.
- Grant PJ. Diabetes mellitus as a prothrombotic condition. J Int Med. 2007;262:157–72.
- Natarajan A, Marshall SM, Worthley SG, Badimon JJ, Zaman AG. The presence of coronary artery disease increases platelet-dependent thrombosis in patients with type 2 diabetes mellitus. J Thromb Haemost. 2008;6:2210–3.
- Trovati M, Anfossi G. Mechanisms involved in platelet hyperactivation and platelet-endothelium interrelationships in diabetes mellitus. Curr Diab Rep. 2002;2:316–22.
